


















Materials Studio软件介绍
Materials Studio 软件介绍
一、Materials Studio 简介
Materials Studio(MS)是一款全尺度材料模拟平台,其拥有优异的操作界面,能够快捷实现模型搭建、参数设定以及结果的可视化分析;MS中融合了多种模拟技术,整合20多个功能模块,实现从电子结构解析到宏观性能预测的全尺度科学模拟研究。历年的更新,MS在功能、效率、精度、使用体验等方面变得更加完善。Perl脚本编写功能使得MS在计算与分析作业变得更加灵活。MS的用户涵盖能源、材料、物理、化学、化工、药物等多个领域,目前为止在国际知名期刊上发表相关的论文超过60,000篇。
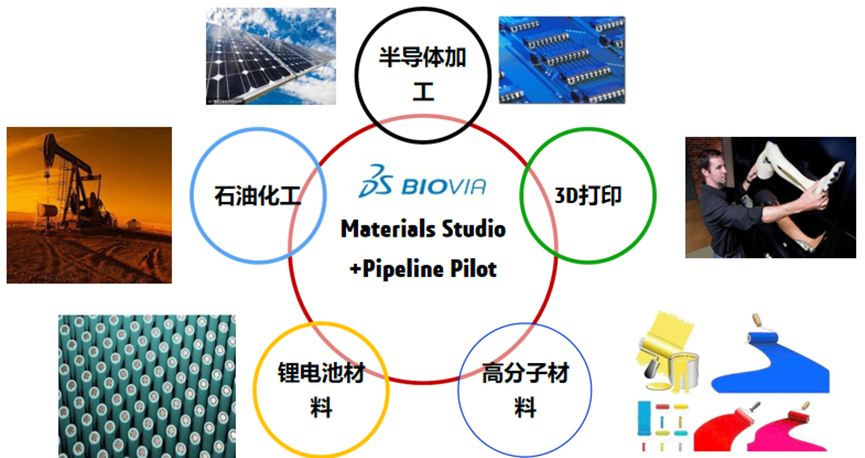
图1 Materials Studio 适用领域
二、Materials Studio的价值
虚拟筛选,节省实际测试与实验的相关成本和时间。通过Materials Studio在计算机中的虚拟世界做实验,可以显著减少实验耗材的使用和节省大量实验时间。
助力创新加速,与实际测试和实验相比,能够更快地开发性能更优异、可持续性更强且成本更低的材料。当材料设计引入分子模拟,可以极快的实现材料开发循环的快速迭代,从而加速材料的开发上市,且更易开发出性能优异的材料。
加深理解,从根本上加深对原子和分子结构与材料属性和行为间关系的理解。材料微观结构决定其性能,基于Materials Studio的分子模拟,可以深入了解材料分原子层级作用机理,极大加深科研工作者对所研究材料的理解。
材料信息学,一体化整合实体实验与计算材料学。
高通量计算和知识管理,通过BIOVIA Pipeline Pilot中的Materials Studio Collection和MaterialsScript API 落实领域最佳实践。
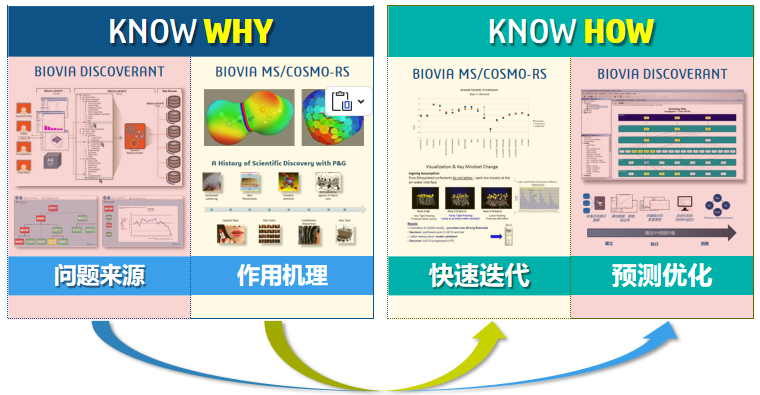
图2 Materials Studio 核心价值概览
三、Materials Studio 软件功能模块
Materials Studio内置量子力学,分子力学,介观模拟,机器学习,统计分析和结晶学等门类齐全的仿真功能。它拥有丰富多样的不同尺度仿真能力,让研究人员能够在各种粒子大小尺度和时间尺度预测材料性能,有效平衡计算时间与计算效率。
操作界面 | 量子力学模块 | 经典模拟模块 |
Visualizer 操作界面 | CASTEP 平面波-赝势密度泛函工具 DMol3 原子轨道线性组合-密度泛函工具 NMR CASTEP 核磁计算工具 CANTERA 化学反应速率方程求解器 QMERA 量子力学与分子力学杂化工具 ONETEP 线性标度密度泛函工具 DFTB+ 紧束缚半经验密度泛函工具 VAMP 半经验分子轨道方法工具 | Amorphous Cell 无定型模型搭建工具 Forcite Plus 分子力学动力学方法 COMPASS III 高精度力场 GULP 专用力场分子力学工具 Conformers 构象搜索和分析 Adsorption Locator 吸附构象搜索工具 Sorption 预测吸收和分离现象 Blends 估算二元体系相容性工具 |
介观模拟模块 | 分析和结晶 | 统计学模块 |
Mesocite 粗粒化模型 MesoDyn 动态平均场密度泛函方法 | Morphology 预测晶体生长形貌 Polymorph Predictor 预测分子晶体堆积和晶形 Reflex 模拟XRD谱图 Reflex Plus 谱图精修 Reflex QPA 分析相组成 X-Cell 谱图精修算法 | QSAR Plus 构效定量关系 Synthia 聚合物种类和共聚物配比筛选工具 |
3.1 Visualizer 操作界面
Materials Visualizer 是材料虚拟建模和仿真领域易用和完善的图形化用户环境,能够帮助化学家、高分子科学家和其他材料科学家提高工作效率,减轻工作量。Materials Visualizer 提供构建、操作和查看分子、晶体材料、表面结构、聚合物和介观结构模型的能力。此外,它还完全支持Materials Studio 的各种仿真结果查看,并能通过图像、动画、图形、图表、表格和文本数据可视化结果。Materials Visualizer 中的大部分工具可通过MaterialsScript API 调用,以便专家级用户创建自定义工具并实现重复性工作自动化。Materials Visualizer 的 Microsoft Windows客户端能配合多种 Windows 和 Linux 服务器运行,为用户提供即时的使用体验。
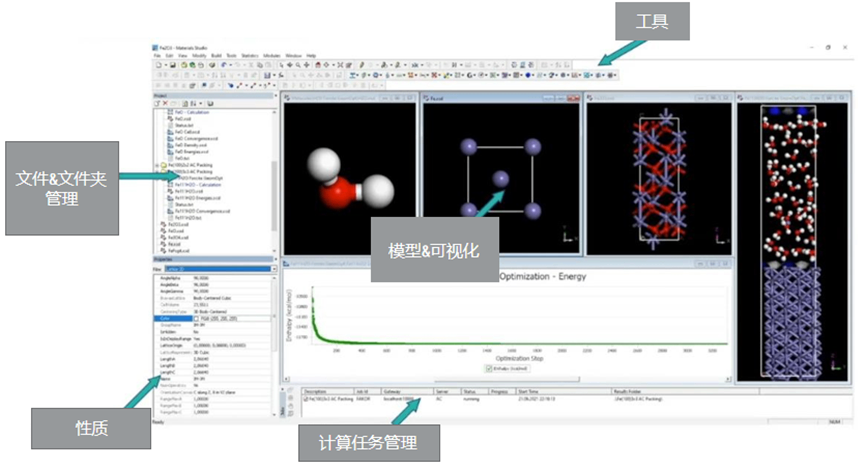
图3 Materials Studio Visualizer 界面展示
3.2 量子力学方法
3.2.1 CASTEP
CASTEP是由剑桥大学研究组开发的一款基于密度泛函理论的先进量子力学程序。程序采用平面波函数描述电子状态,利用赝势替代内层电子,也被称为平面波赝势方法。适于解决固体物理、材料物理、材料科学、化学以及化工等领域中的各类问题。所涉及的研究对象包括半导体、陶瓷、金属、分子筛等各类晶体材料,以及掺杂、位错、界面、表面等各种缺陷结构。
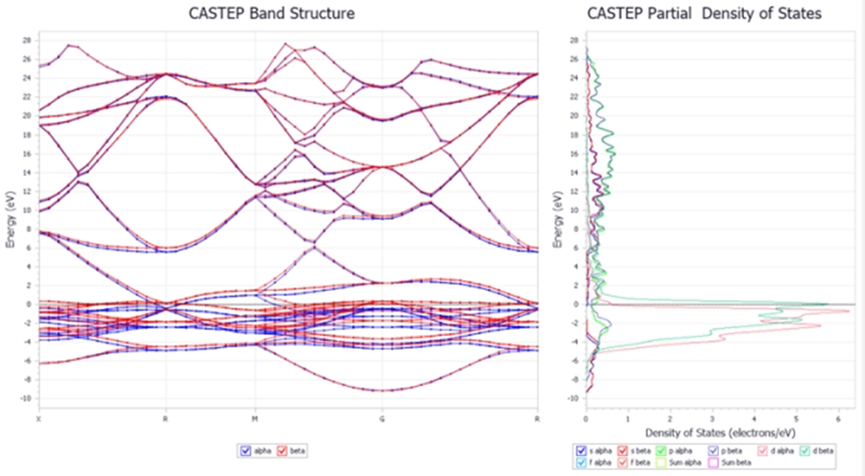
图4 CASTEP 模块计算能带和态密度
3.2.2 DMol3
DMol3采用原子轨道线性组合的方法描述体系的电子状态。该方式兼顾了计算精度和效率,使得DMol3成为一款高效实用的量子力学程序。可以预测材料的电子结构、光学、力学、热力学性能以及气相、溶液、表面及其它固态环境中的化学反应,适合解决化学、化工、生物、材料、物理等领域中的各类问题,尤其是化学反应机理及催化领域的问题。
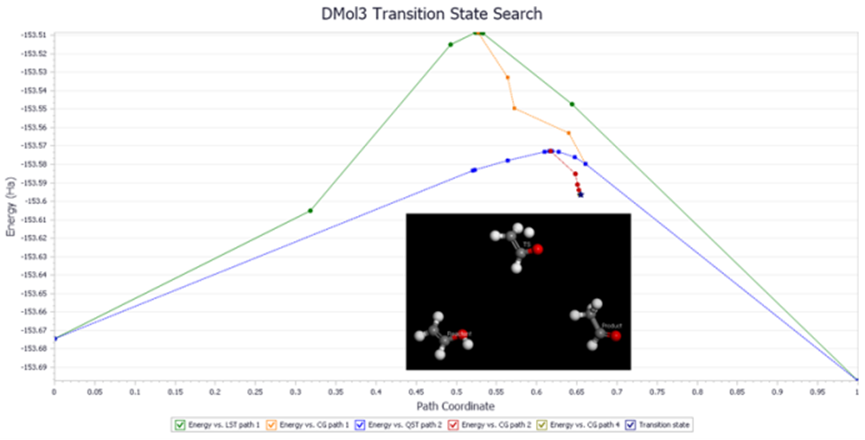
图5 DMol3 模块进行过渡态搜索
3.2.3 NMR CASTEP
NMR CASTEP 依据第一性原理模拟 NMR 化学位移和电场梯度张量。能够进行分子核磁和固体核磁的计算,适用于陶瓷和半导体等多种材料体系。
3.2.4 CANTERA
Cantera [www.cantera.org]是一种化学反应速率方程求解器。Materials Studio Cantera 为化学反应速率的求解提供热力学输入和运行环境。通过Cantera Reaction Editor,用户能够创建新的反应物种和反应类型,并基于Materials Studio DMol3 获取反应相关信息,并将反应类型、物种、反应速率等信息加入已有反应机理文件中。
3.2.5 QMERA
QMERA是一款将量子力学方法的精确性与经典模拟方法的高效性有机结合的程序,在计算的模型中划分量子力学区域和分子力学区域;然后分别调用DMol3模块和GULP模块进行处里。QMERA 提供了多种方式解决两个区域间的耦合问题。它可研究包含上千个原子的体系,在充分考虑周围原子影响的条件下,得到其核心部分的电子结构、可能的化学反应机理、紫外已见光谱、红外光谱等信息。这一方法在非均相催化、表界面吸附、聚合物间的相互作用、生物分子活性的研究中相比于传统量化方法更具优势。
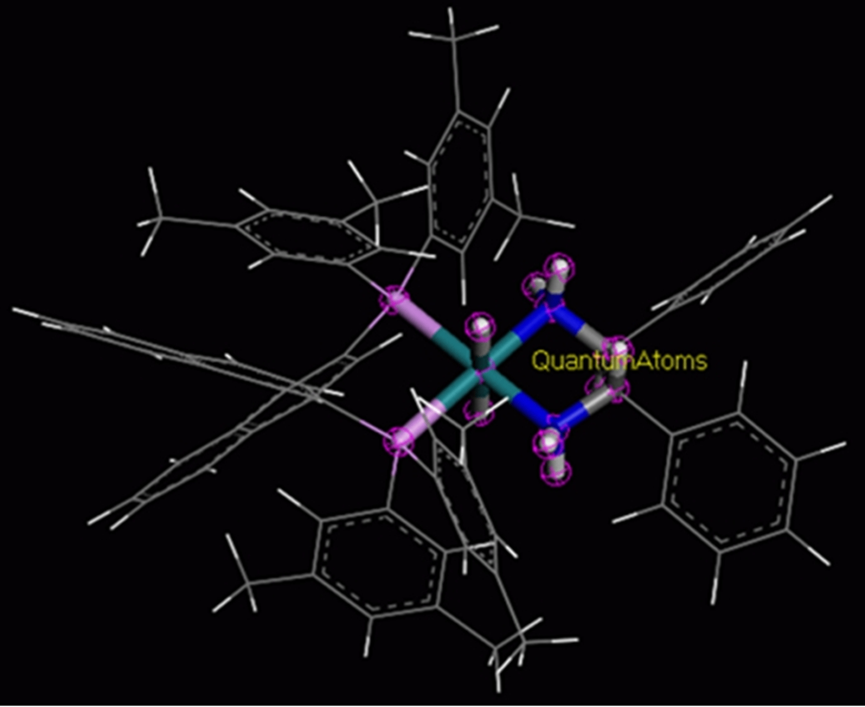
图6 QMERA 在结构中划分量子力学区域和分子力学区域
3.2.6 ONETEP
ONETEP是由剑桥大学研究组开发的一款专门针对大体系(>500原子)的量子力学程序。其关键技术是采用非正交的广义万尼尔(Wannier)函数替代平面波函数进行计算,使模拟计算的时间与体系的大小成线性关系。ONETEP 也被称为线性标度的量子力学方法。可以计算 体系电子态密度、电子局域函数、光学性质、电子激发态、轨道、布局;其应用范围主要包括表面化学、大分子体系(蛋白质、DNA、抗体)及其它复合材料、纳米材料以及半导体、陶瓷材料缺陷等。
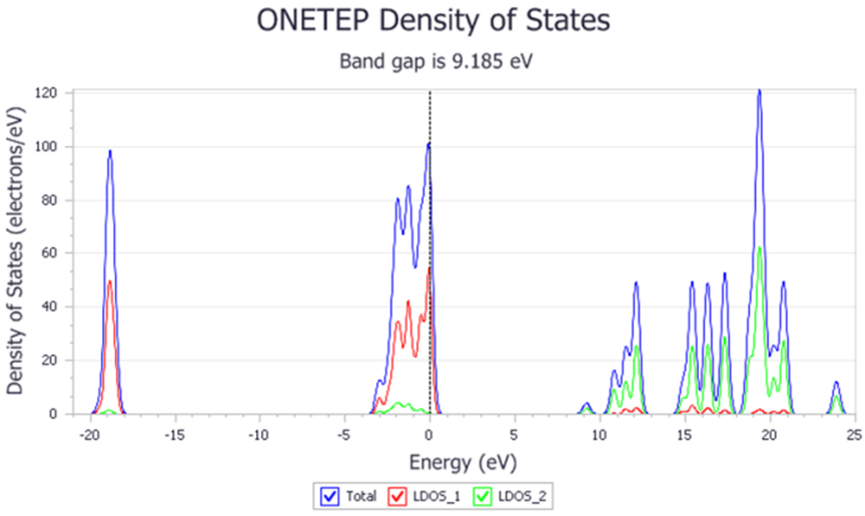
图7 ONETEP计算态密度
3.2.7 DFTB+
DFTB+是一款融合了密度泛函方法(DFT)准确性和紧束缚方法(TB)高效性的半经验量子力学程序,其中所采用的原子轨道波函数和原子核间相互作用势均基于DMol3的结果拟合得到。DFTB+可以对数千个原子体系进行模拟研究,为解决电子、催化、化学化工等领域中各种问题的模拟方法。对于传统量化计算反应动力学计算量方便的问题,DFTB+有其独有的优势。所涉及的研究对象包括有机分子、团簇、绝缘体、半导体、金属,甚至是生物大分子等各类非周期性和周期性体系。
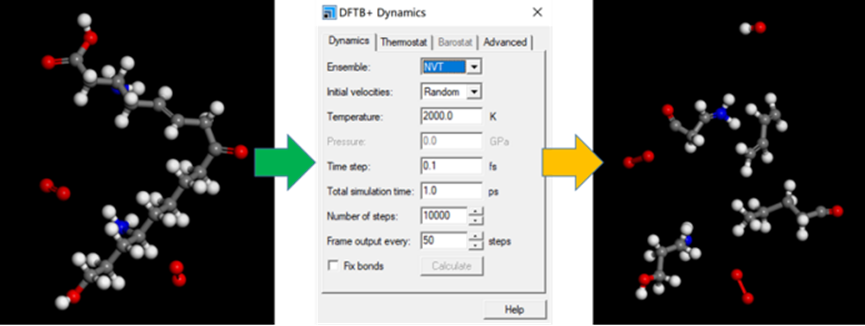
图8 DFTB+ 做动力学计算
3.2.8 VAMP
VAMP是一款基于原子轨道线性组合方法的半经验量子力学程序。它通过忽略部分不太重要的原子轨道重叠积分或者用经验参数替代部分轨道重叠积分的方式简化计算。可以计算非周期体系的电子密度、静电势、紫外可见光谱、熵焓热力学性能、轨道等。VAMP 主要是对有机和无机分子体系进行模拟计算,它可以快速计算多种物理和化学性质。
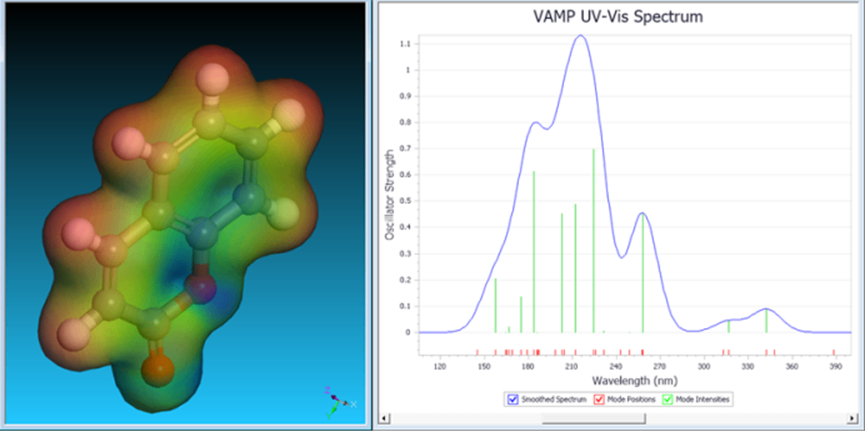
图9 VAMP紫外可见光谱计算
3.3 分子力学动力学模块
3.3.1 Amorphous Cell
Amorphous Cell模块是一个采用蒙特卡洛方法搭建无定形模型的工具。它可用于搭建具有多种分及不同配比的共混模型、溶液模型、复合材料模型、固液/固气界面模型、孔道填充模型等。
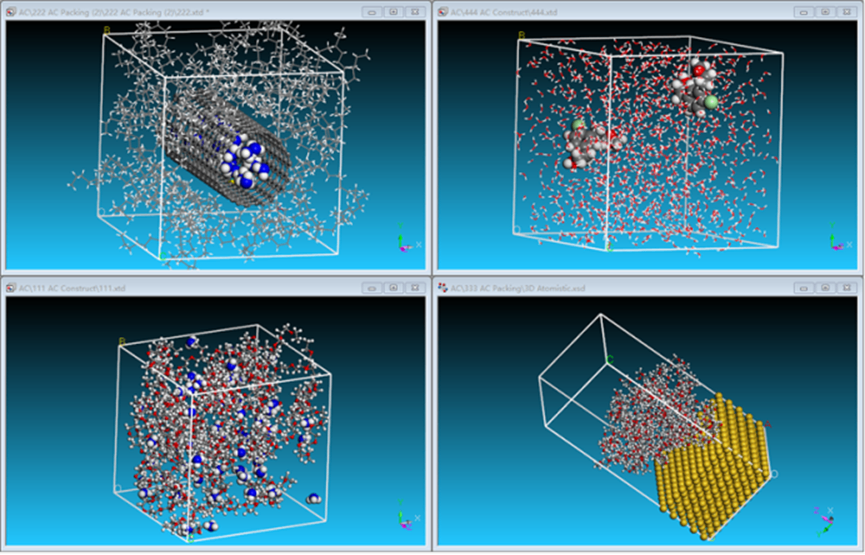
图10 AC模块构建无定型晶胞模型
3.3.2 Forcite Plus
Forcite Plus 是一款分子力学和分子动力学模拟程序。使用力场方法对聚合物、溶液、金属等体系在不同温度压力条件下的热力学、动力学、力学等性能。支持PCFF、CVFF、UFF、Dreiding、COMPASS、DaiKongLiLiu、FinnisSinclair、SuttonChen、ZhouJohnsonWadley等力场。计算任务有:能量计算、几何优化、动力学、剪切/限制性剪切、力学性能、溶解自由能;支持GPU计算。
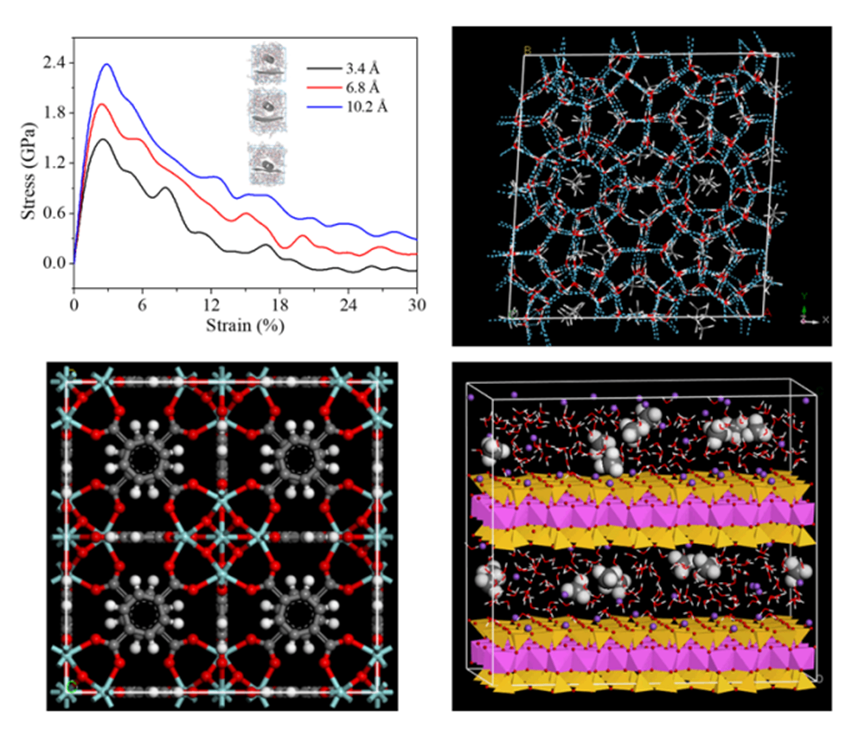
图11 Forcite Plus应力应变曲线计算
3.3.3 COMPASS
COMPASS是一个强大的力场,支持凝聚态材料的原子模拟。COMPASS是第一个使用凝聚相特性参数化和验证的从头算力场,此外还有各种孤立分子的从头算和经验数据。COMPASS可在一个很大的温度、压力范围内,精确地预测多种单分子及其凝聚态的结构、构象、振动及热物理性质。包含对离子液体、杂环分子等特殊体系的支持,目COMPASS版本已更新至COMPASSIII。
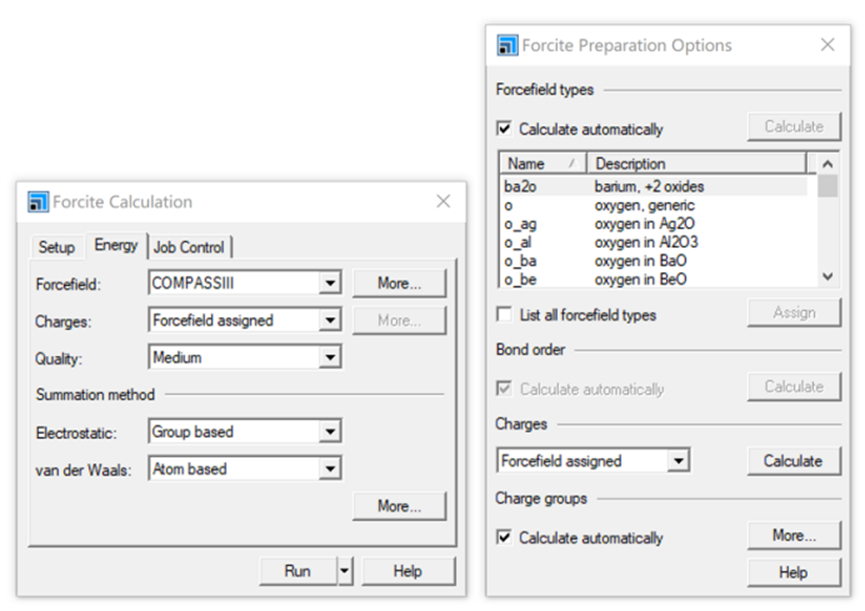
图12 COMPASS 力场设置界面
3.3.4 GULP
GULP是一款分子力学和分子动力学模拟程序。它可以对具有零维、一维、二维、三维结构的各种材料体系的多种性质进行计算。GULP 具有多种针对性较强的势函数,警如支持壳层模型的 Bush、Lewis 势以及多种原子嵌入势(EAM)和改良嵌入势(MEAM),针对粘土矿物的ClayFF、针对碳材料的Brenner、Tersof、针对化学反应的ReaxFF;同时GULP 提供了拟合和编辑势函数的工具,提高模拟计算精度。
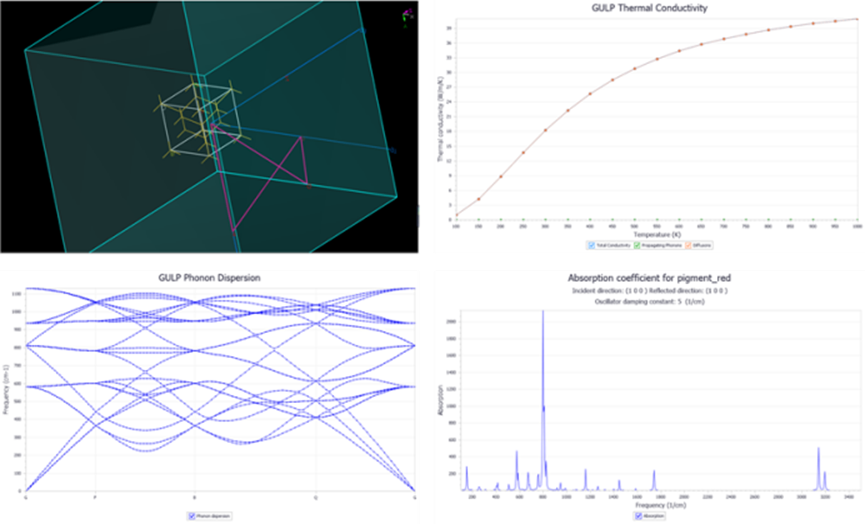
图13 GULP的分子动力学计算
3.3.5 Conformers
Conformers 是一款以分子力学为基础,提供了搜索非周期分子体系的构象空间的方法,以获得合理的低能构象抽样。研究的主要自由度是分子体系的可旋转扭转角的集合。并且具有一定的分析功能,可以建立分子构象与其能量、偶极矩、回转半径之间的关系。
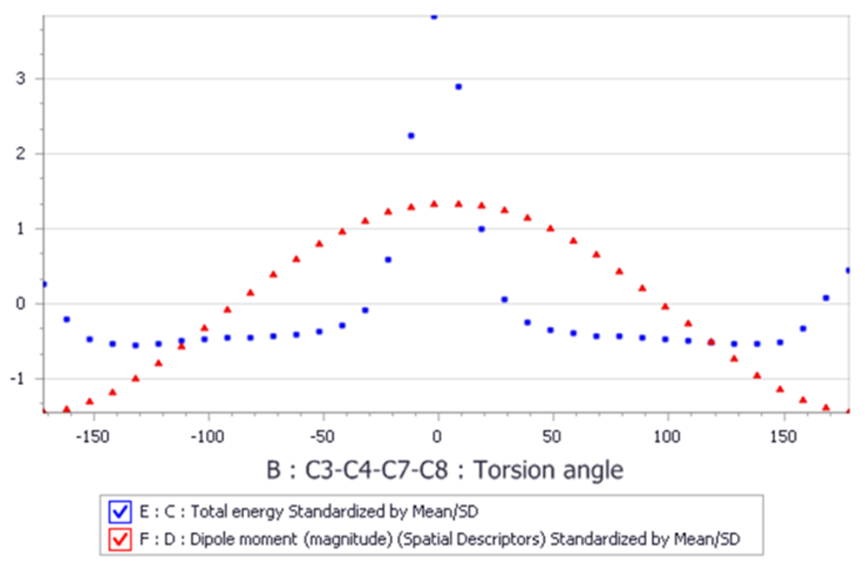
图14 Conformers构像搜索
3.3.6 Adsorption Locator
Adsorption Locator是一款采用蒙特卡罗退火方法搜索吸附质在吸附剂材料上的最稳定吸附构象的程序,它可以获得吸附剂上吸附位点、吸附质的稳定吸附构象。在表面腐蚀、催化剂设计以及晶体结晶形貌等领域具有理论指导意义。
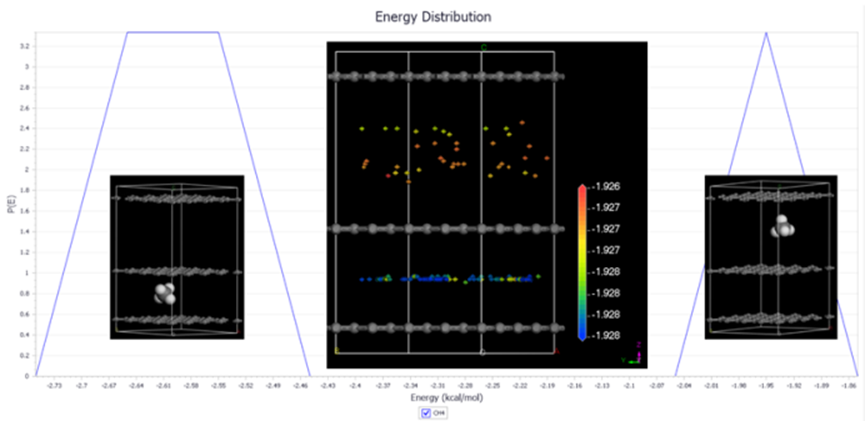
图15 Adsorption Locator 最稳定吸附构像搜索
3.3.7 Sorption
Sorption是一款基于巨正则蒙特卡洛(GCMC)方法预测单一或混合组分在微孔材料和介孔材料中吸附的程序。多孔材料包括分子筛、硅铝酸盐、粘土、纳米管、聚合物膜、活性炭、COF、MOF等材料;Sorptior可直接给出吸附等温线、吸附量、吸附热、亨利常数等性质,可应用于催化、气体分离、气体传感器以及离子交换等诸多研究领域。
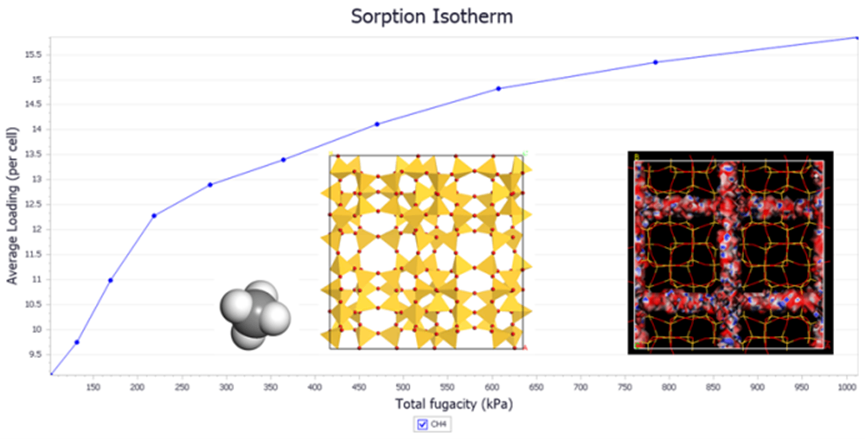
图16 Sorption 空隙材料吸附研究
3.3.8 Blends
Blends 是一款以力场为基础,采用扩展的 Flory-Huggins 模型估算二元混合物体系相容性的程序,可以有效的缩短工艺探索周期。这些二元混合物包括溶剂-溶剂、聚合物 -溶剂以及聚合物-聚合物。这种模拟技术能够直接从二元混合物的化学结构预测出混合物的热力学性质。作为一个快速的筛选工具,Blends 可以在缩减试验次数的同时开发出稳定的产品配方,它在粘结剂、医药、化妆品等材料制备领域具有重要作用。
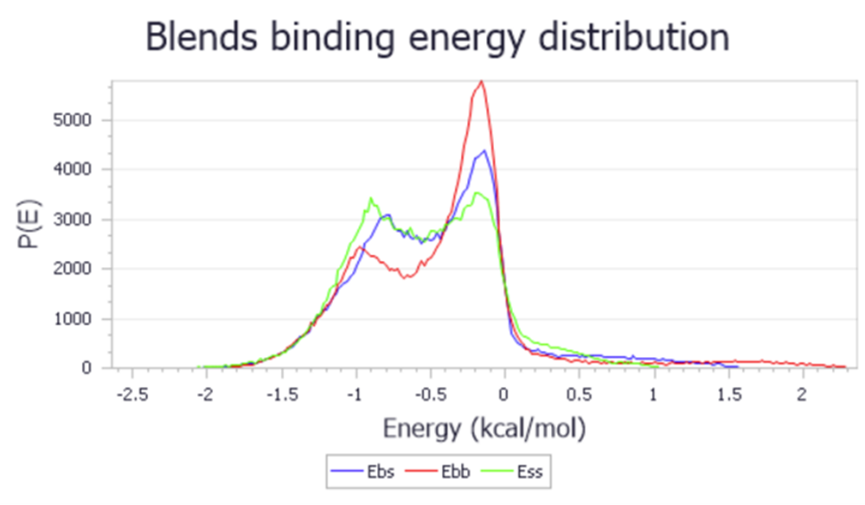
图17 Blends 混合体系能量研究
3.4 介观模拟
3.4.1 Mesocite
Mesocite 是一个包含粗粒化分子动力学(CGMD)、耗散粒子动力学(DPD)和颗粒动力学(Granular Dynamics)方法,含有Martini、Martini 3、Shinoda2007力场。其中Martini 3的参数化将中尺度力场的能力扩展到各种系统。这些系统可以是跨膜蛋白质到聚合物膜、大纳米结构以及小分子结构到离子构型的膜材料。考虑到每个粒子的化学性质,Martini 3力场引入更多的力场类型和中尺度粒子(珠子)之间的范德华相互作用。与之前的版本相比,Martini 3功能更加强大。并以软凝聚态材料为主要研究对象的介观模拟工具依靠介观方法在时间和空间尺度上的优势,Mesocite 可以更加快捷的研究添加剂、溶剂、单体类型、组分比例对各种均聚、嵌段枝状聚合物结构、热力学性能、扩散等性能的影响;在复合材料、涂料、电池电极材料制备、化妆品以及药物的控释领域具有重要应用。该模块支持GPU计算。
图18 Mesocite 颗粒动力学研究
3.4.2 Mesodyn
MesoDyn是一款基于动态平均场密度泛函方法的介观模拟程序,主要用于复杂流体,包括聚合物熔体和混合体系在介观尺度的动力学研究。MesoDyn可以模拟100-1000 nm的体系,可以非常方便的研究复杂流体、聚合物共混的动力学过程和稳定拓扑形貌,在涂料、化妆品、混合材料、表面活性剂、复杂药物传输以及相关领域具有广泛应用。
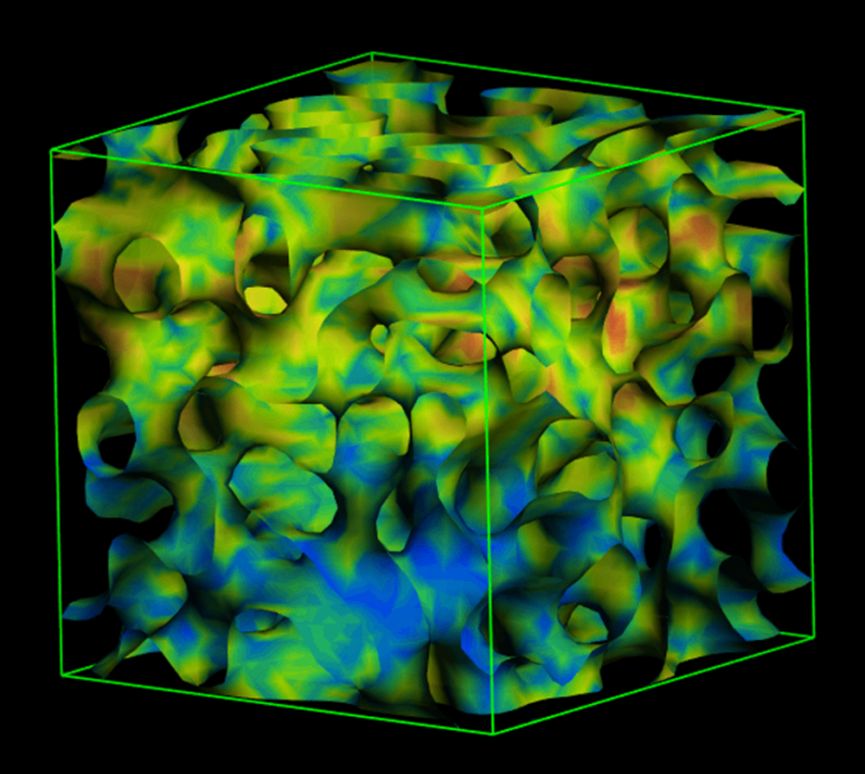
图19 MesoDyn 动态平均场密度结构图
3.5 晶体晶型预测、形貌与结构解析
3.5.1 Morphlogoy
Morphology 是一个通过材料晶体结构预测其晶粒形貌的工具。它可对特定添加剂、溶剂以及杂质存在下的晶体形貌研究提供帮助。其主要应用领域包括医药品、农用化学品、食品科学石油化工、水泥、日用品以及特殊化学品等。
3.5.2 Polymorph
Polymorph Predictor是一个以力场为基础,采用蒙特卡洛模拟退火法,通过直接利用化合物的分子结构预测其多型。获得晶型与密度等相关数据。晶体材料在制药、农药、染料、炸药以及专用化学品工业中有着非常普遍的应用。
3.5.3 Reflex Plus
Reflex可以模拟x射线,电子和中子衍射数据,提供了从有机、无机、有机金属和生物晶体的衍射模式中提取最大量信息所需的所有工具。粉末定量相分析(QPA)用于根据混合物的粉末模式,以及纯相的结构模型或粉末模式,确定相混合物中不同相的相对量。包含多种指标化方法,ton通过实验XRD数据获得晶胞参数和空间群等信息。Reflex还可以考虑晶体的能量,在这种新颖的Pawley方法中,实验模式和模拟模式之间的相似性与势能共同优化,从而产生既与实验模式匹配又具有接近最小势能的晶体结构。
其中Reflex 用于根据晶体材料模型仿真X 射线、中子和电子粉末衍射谱图。Reflex Plus 为根据中高质量粉末衍射数据判定晶体结构提供了一整套完整软件包。Reflex QPA 扩展了Reflex 的功能,能够进行物相分析,可根据粉末衍射数据,确定混合物中各个单相(包括无机系统和有机系统)的相对比例。
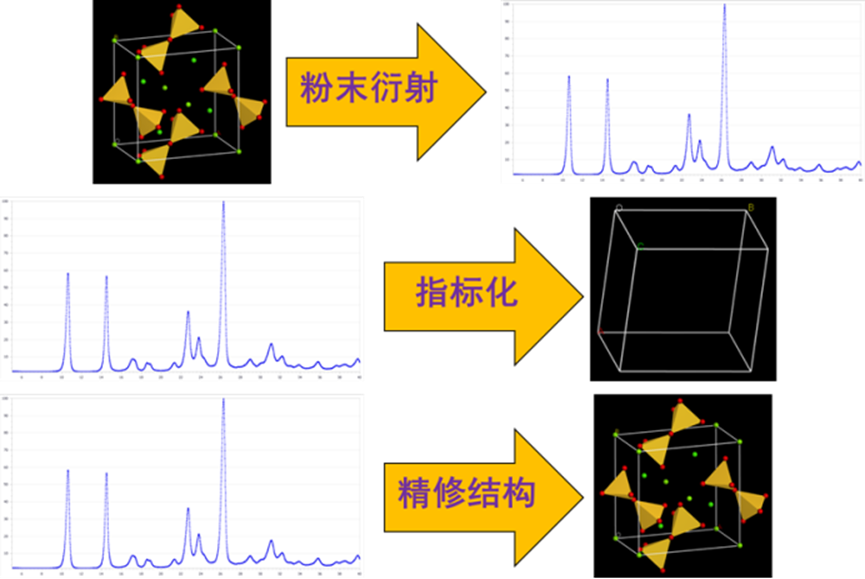
图20 Reflex x射线模拟
3.5.4 X-Cell
X-Cell 是一种用于中高质量粉末衍射数据的高效率索引算法。X-Cell 使用优化的两分法流程穷举搜索参数空间,为所有可能的单位晶格解建立完整列表。
3.6 构效关系
3.6.1 QSAR
QSAR(Quantitative Structure Activity Relationship,定量构效关系)模块用于在实验信息(“活性”)和分子水平特征(“描述符”)之间创建统计回归模型。该方法需要一组具有已知属性的材料数据。然后可以计算分子描述符,并生成将属性与描述符相关的数学回归模型。然后可以用这个数学模型来预测具有未知活性的材料。也可以包括加工条件和配方数据的描述符。QSAR的应用可以极大的提升高性能材料的研发速率。对于描述符的获取可以调用Forcite、VAMP、DMol3等模块计算。
图21 QSAR模型
3.6.2 Synthia
Synthia 是一款以美国陶氏公司Jozef Bicerano 博士的工作为基础,使用聚合物的拓信息,建立原子和化学键以及聚合物性质的关联,通过定量结构-性能关系方法预测聚合物热力学、力学和输运性质的程序; Synthia 使用,包含碳、氢、氮、氧、硅、硫、氟,氯、溴9中元素组成的聚合物的体系均可预测;Synthia 的预测结果在实际工作中得到了广泛的验证。
图22 Synthia 计算界面
3.7 Perl Script脚本功能
MaterialsScript应用程序编程(API)接口是一个对象模型,Perl等脚本语言可以使用它来控制Materials Studio和Materials Server。实现批量化自动计算、批量化数据处理以及体系之间的化学反应,如氢键类型统计、材料拉伸、反应动力学产物统计、SEI膜生长、环氧树脂体系交联等。
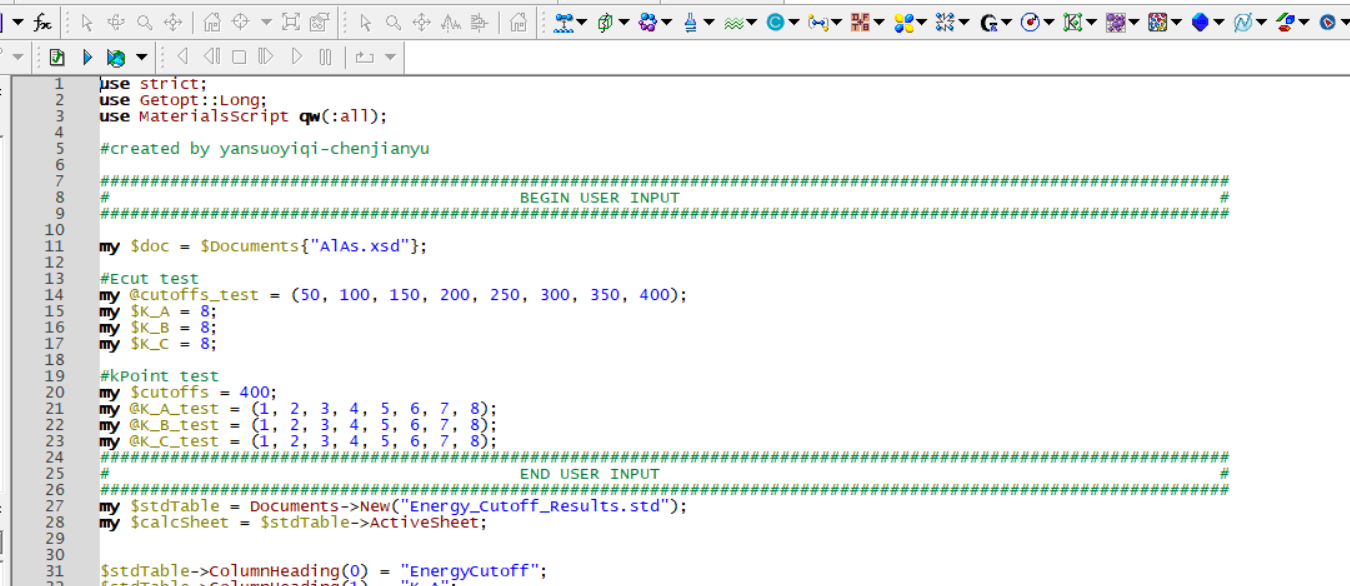
图23 Materials Studio中自洽收敛性测试脚本
四、Materials Studio软件架构
Materials Studio软件采用灵活的Client-Server结构。其核心模块Visualizer运行于客户端PC,支持的操作系统主要为Windows系列;计算模块(如Forcite,Amorphous,DMol3,CASTEP等)运行于服务器端,支持Windows系统以及Linux等多种操作平台。浮动许可(Floating License)机制允许用户将计算作业提交到网络上的任何一台服务器上,并将结果返回到客户端进行分析,从而最大限度地利用了网络资源。
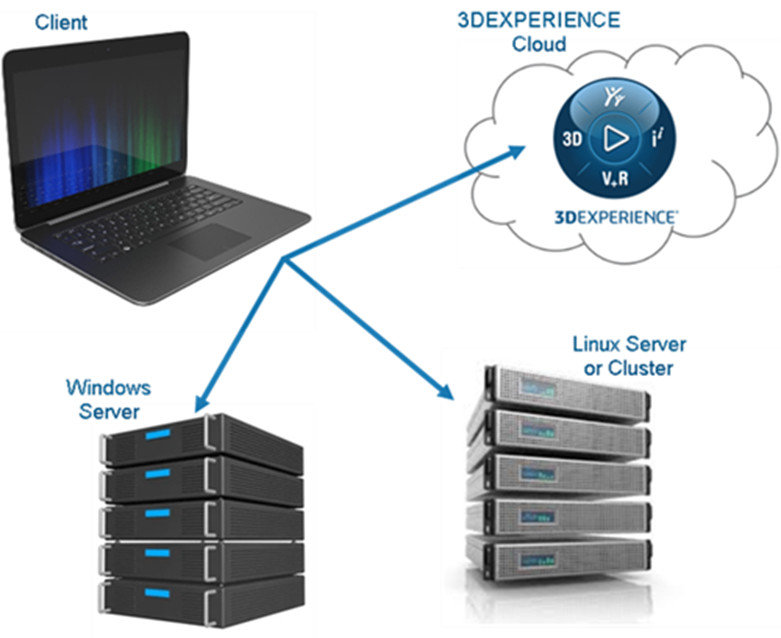
图24 Materials Studio部署架构